

Was ist die ISO 13485 für das Qualitätsmanagement Medizinprodukte?
Die ISO 13485 enthält Anforderungen an das Qualitätsmanagement für Medizinprodukte. Dieses soll die Qualität medizinischer Produkte erhalten und verbessern und somit die Sicherheit der Nutzer sicherstellen. Im Qualitätsmanagement werden die Prozesse optimiert und potenzielle Sicherheitsrisiken für das Medizinprodukt aufgedeckt sowie reduziert. Das Qualitätsmanagementsystem (QMS) überprüft die Erfüllung normativer Forderungen sowie damit einhergehend gesetzlicher Anforderungen auf deren Erfüllung. Dies ist selbstverständlich eine Grundvoraussetzung, damit Unternehmen Medizinprodukte überhaupt anbieten und verkaufen dürfen.
Die Norm ISO 13485 ist für Hersteller von Medizinprodukten relevant, die nationale, europäische sowie internationale Regelungen erfüllen müssen. Es ist ebenso möglich, dass Lieferanten von Medical Devices sowie externe Dienstleister ein QM-System oder sogar eine Zertifizierung nach ISO 13485 nachweisen müssen. Durch die Umsetzung der Normforderungen zeigen Firmen, dass sie den Lebenszyklus von Medizinprodukten sowie zugehörige Tätigkeiten ständig leiten und lenken können. Es wird zudem sichergestellt, dass die Risiken bei der Herstellung, dem Vertrieb oder der Anwendung von medizinischen Produkten möglichst minimiert werden.
Was ist die Definition von Qualitätsmanagement für Medizinprodukte?
Qualitätsmanagement bzw. ein Qualitätsmanagementsystem beschreibt die geplante, systematische Herangehensweise an die Verbesserung der betrieblichen Prozesse und Produkt- bzw. Dienstleistungsqualität anhand normativer Vorgaben sowie Kundenwünschen (Anforderungen) durch das Schaffen angemessener Rahmenbedingungen mit einer angemessenen Aufbau- und Ablauforganisation sowie deren ständige Verbesserung. Qualitätsmanagement Medizinprodukte bezeichnet folglich dieses Vorgehen für Unternehmen, die im Bereich Medizinprodukte (Medizinproduktehersteller) tätig sind. Hier sind sowohl die normativen als auch die gesetzlichen Rahmenbedingungen weitaus strikter.
Alle unsere YouTube Videos finden Sie hier auf dem YouTube Kanal der VOREST AG!
Wie hängt die ISO 13485 mit der ISO 9001 zusammen?
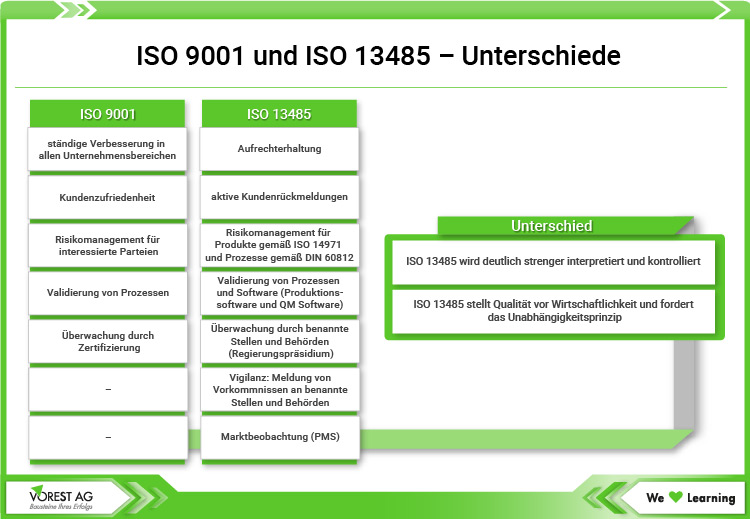
Die ISO 13485 ist zwar eine eigenständige Norm für Qualitätsmanagement im Bereich Medizinprodukte, basiert aber dennoch auf der ISO 9001. Anders als die Qualitätsmanagement Norm ISO 9001:2015 folgt die Norm für Medical Devices jedoch nicht der High Level Structure. Zudem stellt die ISO 13485 neben den Kundenanforderungen auch die Produktanforderungen, die im Zusammenhang mit der Sicherheit und Leistung stehen, in den Vordergrund.
Zwar ist ein Großteil der Forderungen bei beiden Normen gleich, jedoch schließt die ISO 13485 einige Forderungen der QM Norm ISO 9001 auch aus. Grund hierfür ist, dass die betroffenen Forderungen nicht als regulatorische Anforderungen geeignet sind. Dennoch können Unternehmen, die bereits nach ISO 9001 zertifiziert sind, auch die Qualitätsmanagementnorm für Medizinprodukte ohne großen Mehraufwand umsetzen. Mehr dazu und eine Gegenüberstellung der Inhalte beider Normen finden Sie in der Herausgabe der ISO 13485 der ISO.
Welche Ziele hat das Qualitätsmanagement Medizinprodukte nach ISO 13485?
Das Medizinprodukte Qualitätsmanagement nach ISO 13485 hat das Ziel, die Qualität medizinischer Produkte zu erhalten und zu verbessern. Ein funktionierendes Qualitätsmanagement ist dabei besonders in der Medizinprodukteindustrie unerlässlich. Denn um die Sicherheit der Patienten und Anwender zu gewährleisten, müssen Medizinprodukte besonders zuverlässig und sicher sein. Hersteller müssen daher eventuelle Sicherheitsrisiken ihrer Produkte kennen und diese auf ein Minimum reduzieren. Auch zahlreiche regulatorische sowie normative Anforderungen müssen erfüllt werden, damit Unternehmen Medizinprodukte überhaupt anbieten und verkaufen dürfen. Hier kommt das Qualitätsmanagementsystem zum Einsatz. Ein solches erfüllt die relevanten Anforderungen und optimiert die Prozesse im Unternehmen soweit, dass Fehler sowie Risiken weitgehend vermieden werden.
Welche Ausbildung benötigen Sie zur Einführung oder Betreuung Ihres Qualitätsmanagement Systems in der Medizinprodukteindustrie?
Mit unseren Schulungen zum Qualitätsmanagement für Medizinprodukte nach DIN ISO 13485 machen wir Sie fit für die Einführung oder laufende Betreuung und Weiterentwicklung Ihres Qualitätsmanagementsystems. Mit unserer modularen Qualifizierung können Sie mit dem Lehrgang Basiswissen ISO 13485 in die Grundlagen der Norm einsteigen und sich anschließend zum Internen Auditor ISO 13485 oder Managementbeauftragten für das Qualitätsmanagement in der Medizinprodukteindustrie ausbilden. Hier finden Sie eine Übersicht zu unserem Gesamtangebot im Bereich ISO 13485.
Kursvideo
Schritt
Seminartitel
Kursformen
Zertifikat
Informationen








Wie ist die Norm DIN EN ISO 13485 für das Medizinprodukte Qualitätsmanagement aufgebaut?
Die ISO 13485 ist in mehrere Hauptkapitel unterteilt, die spezifische Anforderungen an das Qualitätsmanagementsystem eines Unternehmens definieren. Die ersten 3 Kapitel diesen als Einleitung. Im Wesentlichen besteht die Norm aus den fünf darauffolgenden Hauptabschnitten 4 bis 8. In diesen Abschnitten stehen die regulatorischen Anforderungen, welche die Sicherheit von Medizinprodukten gewährleisten sollen, im Fokus. Neben den Kundenanforderungen geht es also um die Produktanforderungen, die im Zusammenhang mit Sicherheit und Leistung stehen. Im Folgenden erläutern wir den Inhalt der ISO 13485 auf Basis der einzelnen Abschnitte.
Abschnitt 1 - Anwendungsbereich der ISO 13485
Dieser Abschnitt definiert, für welche Unternehmen und Aktivitäten die Norm anwendbar ist. Sie umfasst alle Organisationen, die an der Entwicklung, Herstellung sowie Bereitstellung von Medizinprodukten beteiligt sind.
Abschnitt 2 - Normative Verweisungen
Hier werden die relevanten Dokumente genannt, auf die in der Norm verwiesen wird. Diese Dokumente sind wichtig, um bestimmte technische oder regulatorische Anforderungen zu verstehen und umzusetzen.
Abschnitt 3 - Begriffe und Definitionen
In diesem Kapitel werden die zentralen Begriffe und Definitionen festgelegt, um sicherzustellen, dass alle beteiligten Parteien die Begrifflichkeiten einheitlich verstehen und anwenden.
Abschnitt 4 - Anforderungen an das Qualitätsmanagementsystem nach ISO 13485
Abschnitt 4 der Norm ist untergliedert in zwei Unterabschnitte. In Abschnitt 4.1 „Allgemeine Anforderungen“ liegt der Fokus auf dem eigenen Unternehmen und dessen Prozesse. Die Norm fordert die Anwendung eines risikobasierten Ansatzes zur Lenkung der geeigneten Prozesse, die für Qualitätsmanagementsysteme benötigt werden. Für jeden Prozess, der das Qualitätsmanagement der Medizinprodukte betrifft, müssen also Kriterien sowie Methoden für dessen Durchführung und Lenkung festgelegt werden. In diesem Zusammenhang muss das Unternehmen die Rollen der Organisation dokumentieren. Zu den Rollen, die von der Organisation übernommen werden, können der Hersteller, der Bevollmächtigte, der Importeur oder Vertriebspartner zählen.
Der darauffolgende Abschnitt 4.2 der ISO 13485 enthält die Anforderungen an die Dokumentation. Neben der Qualitätspolitik müssen beispielsweise auch die Qualitätsziele dokumentiert werden. Bei der Dokumentation ist die Lenkung der Aufzeichnungen besonders wichtig. Unternehmen müssen Dokumente unter anderem vor ihrer Herausgabe bewerten und genehmigen oder den Verlust durch geeignete Verfahren verhindern. Im Zusammenhang mit der Dokumentation spielt die Medizinprodukteakte eine besondere Rolle. Hersteller von Medizinprodukten müssen diese Akte für jeden Typ oder jede Gruppe von Medizinprodukten erstellen. Dabei umfasst die diese alle Dokumente, welche die Konformität mit den geltenden normativen sowie regulatorischen Anforderungen nachweisen.
Abschnitt 5 - Verantwortung der Leitung
Nachdem die Anforderungen an das Qualitätsmanagement System vom Medizinprodukte Hersteller definiert wurden, geht es in diesem Abschnitt um die Verantwortung der Leitung. Hier werden der obersten Leitung konkret Aufgaben in Bezug auf das QM-System überlassen. Es reicht dabei nicht aus als Geschäftsführer, Vorstand oder oberstes Management die Aufgaben an die Mitarbeiter zu delegieren und sich selbst nicht in das Managementsystem einzubringen. Die Norm ISO 13485 weist der Führung aktive Aufgaben zu. Unter anderem zählen die Entwicklung sowie die Aufrechterhaltung des Managementsystems zu den Aufgaben der obersten Leitung. Diese muss beispielsweise eine Qualitätspolitik und Qualitätsziele definieren und sicherstellen, dass diese den Mitarbeitern bekannt sind.
Weiterhin muss die Führung den Mitarbeitern die Bedeutung der regulatorischen Anforderungen näher bringen. Auch die Festlegung von Verantwortlichkeiten und Befugnissen sind Aufgaben der Führung. Ebenso muss sie einen Beauftragten der Leitung benennen. Hier besteht ein wesentlicher Unterschied zur ISO 9001. In dieser wird die Festlegung eines Qualitätsmanagementbeauftragten (QMB) nämlich nicht mehr explizit gefordert. Eine weitere Aufgabe der obersten Leitung ist die Durchführung der Managementbewertung. Hierbei handelt es sich um die regelmäßige Bewertung der Leistung der Qualitätsmanagementsysteme. Die Führung prüft in diesem Zusammenhang, ob das System auch wie geplant funktioniert und wirksam ist. Die erhaltenen Ergebnisse dienen dann als Basis, um Verbesserungsmaßnahmen abzuleiten.

Passende Blogbeiträge zum Thema:
Entdecken Sie auch unsere Blogbeiträge zum Qualitätsmanagement für Medizinprodukte oder zum Audit und erhalten Sie Expertenwissen unter anderem zu diesen Themen:
Abschnitt 6 - Management von Ressourcen
In diesem Abschnitt der ISO 13485 geht es darum, die notwendigen Ressourcen für die Umsetzung des Qualitätsmanagement Systems für Medizinprodukte bereitzustellen. Auch dies ist eine Aufgabe, die in den Verantwortungsbereich der Leitung fällt. Es geht hier darum, die zeitlichen, monetären und personellen Ressourcen zu planen, festzulegen und bereitzustellen. Organisationen müssen zudem die benötigte Kompetenz der Mitarbeiter, die am QM beteiligt sind, definieren und für ein entsprechendes Qualitätsbewusstsein sorgen. Um die benötigten Kompetenzen zu erreichen und aufrechtzuerhalten, müssen zudem ISO 13485 Schulungen oder andere Maßnahmen geplant werden. Auch die Sicherstellung der Infrastruktur spielt in der ISO 13485 eine wichtige Rolle. So müssen Medizinproduktehersteller dafür sorgen, dass die Infrastruktur die Sicherheit und Leistung der Produkte gewährleistet und Produktverwechselungen vermieden werden. Diese Anforderung kann zum Beispiel durch die räumliche Trennung von Prozessen oder die Beschriftung der Herstellungsräume sichergestellt werden.
Abschnitt 7 - Produktrealisierung
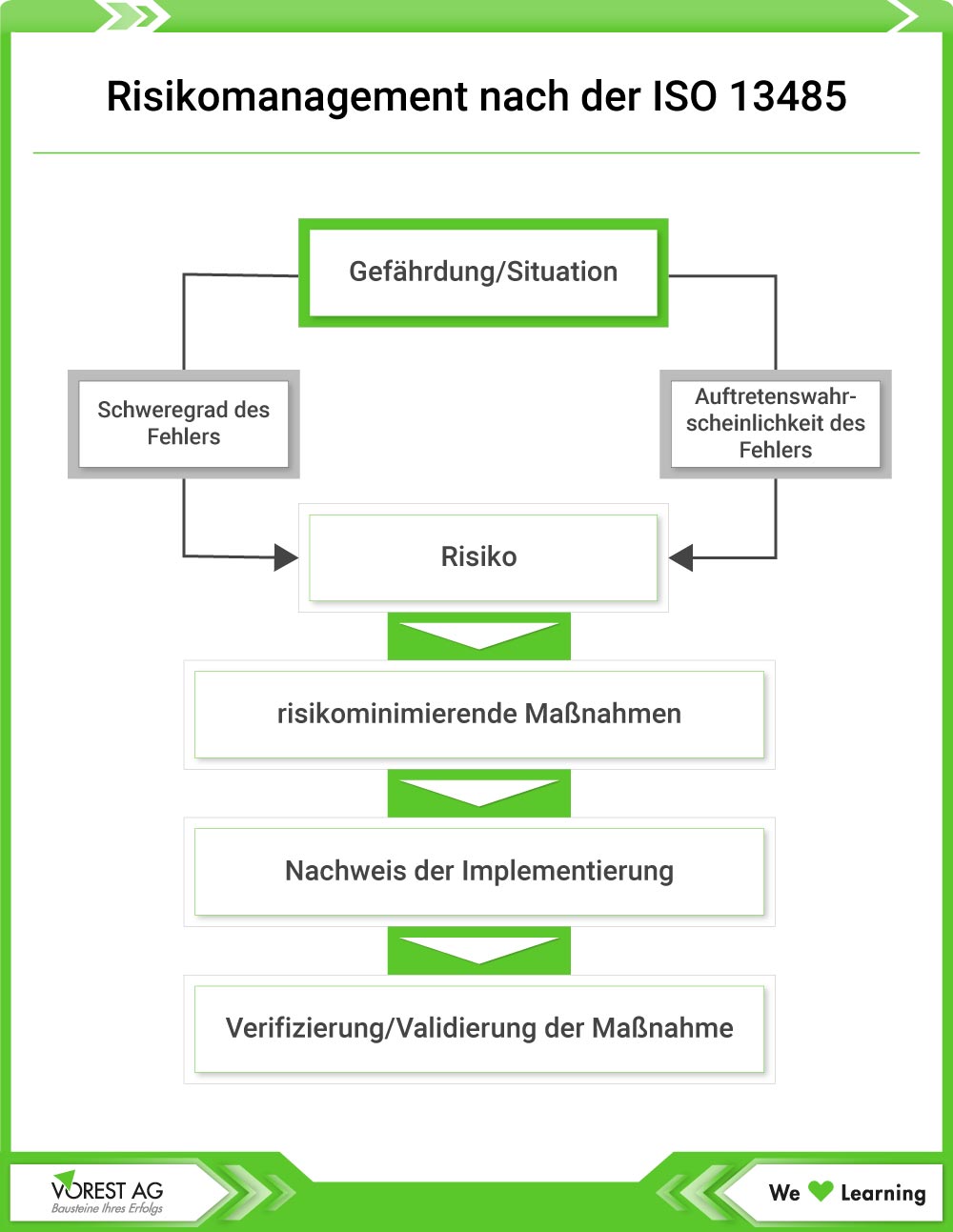
Dieser Abschnitt der ISO 13485 ist sehr umfangreich und umfasst Anforderungen, welche die Herstellung der Medizinprodukte betreffen. Im Mittelpunkt stehen dabei die Prozesse, die für die Planung, Entwicklung von Medizinprodukten und den Verkauf der Produkte relevant sind. Dies betrifft auch das Risikomanagement. Im Unterabschnitt „Kundenbezogene Prozesse“ geht es dann um die Kundenanforderungen. So müssen Hersteller von Medizinprodukten die Anforderungen der Kunden zunächst ermitteln und bewerten. Die Organisation muss zudem jede erforderliche Anwenderschulung zur Sicherstellung der festgelegten Leistung und der sicheren Anwendung des Medizinprodukts ermitteln. Auch eine ausreichende Kommunikation mit den Kunden wird gefordert. Die Anforderungen beziehen sich beispielsweise auf eine ausreichende Produktinformation oder die Rückmeldungen der Kunden.
Die „Entwicklung“ ist ein weiterer Unterabschnitt der Qualitätsmanagement Norm für Medizinprodukte. Die ISO 13485 definiert in diesem Abschnitt, wie der Prozess der Produktentwicklung zu dokumentieren ist. So muss die Entwicklung geplant und gelenkt werden. Unternehmen haben zudem die Pflicht, die Planung auf aktuellem Stand zu halten und in einer Entwicklungsakte zusammenzufassen. Die Dokumentation des Entwicklungsprozesses muss dabei folgende Inhalte umfassen:
- Entwicklungsphasen und deren Bewertung
- Verifizierung, Validierung und Tätigkeiten des Designtransfers
- Entwicklungsteam und weitere Verantwortliche sowie Ressourcen im Projekt
- Sicherstellung der Rückverfolgbarkeit der Entwicklungsergebnisse
Die weiteren Unterabschnitte zur Produktrealisierung befassen sich mit der Beschaffung, der Produktion und Erbringung von Dienstleistungen sowie mit der Lenkung von Überwachungs- und Messmitteln. In der Norm finden Sie zum Beispiel genaue Anforderungen, die die Auswahl und Bewertung der Lieferanten betreffen. Damit ein Produkt gemäß den Spezifikationen erstellt wird, stellt die ISO 13485 unter anderem spezielle Anforderungen an die Sauberkeit. Dies ist vor allem bei sterilen Produkten unerlässlich. Alle Geräte, die bei der Herstellung medizinischer Produkte eingesetzt werden, sind dabei zu überwachen. Zudem müssen Unternehmen diese Geräte regelmäßig kalibrieren und auf ihre Eignung prüfen.
Alle unsere YouTube Videos finden Sie hier auf dem YouTube Kanal der VOREST AG!
Abschnitt 8 - Messung, Analyse und Verbesserung
Im letzten Abschnitt geht es um die kontinuierliche Verbesserung (KVP). Wie auch andere Normen für Managementsysteme stellt die ISO 13485 die Verbesserung in den Fokus. Diese ist notwendig, um das Qualitätsmanagementsystem und dessen Wirksamkeit dauerhaft aufrechtzuerhalten. Ein wichtiges Instrument, dass bei der Verbesserung zum Einsatz kommt, ist das interne Audit. Die Forderung nach einem internen Audit ist im Unterabschnitt 8.2 „Überwachung und Messung“ definiert. Im Rahmen des internen Audits prüft der Auditor, inwieweit das QM System die normativen sowie firmeninternen Anforderungen umsetzt. Die Norm fordert in diesem Zusammenhang die Dokumentation der Planung und Durchführung der Audits. Wichtige Dokumente sind dabei unter anderem eine Audit-Checkliste oder der Auditbericht.
Des Weiteren fordert die ISO 13485 die Lenkung nichtkonformer Produkte. Produzenten medizinischer Produkte müssen durch entsprechende Prozesse vermeiden, dass nichtkonforme Produkte ausgeliefert werden. Ebenso verlangt die Norm eine regelmäßige Datenanalyse. Hier müssen Sie Verfahren zur Ermittlung, Erfassung und zur Analyse geeigneter Daten festhalten. Die geforderte Datenanalyse dient dabei auch zur Prüfung der Wirksamkeit des QM Systems. In diesem Zusammenhang müssen Sie auch Daten einbeziehen, die Sie im Rahmen der Überwachung und Messung aus anderen Quellen gewonnen haben:
- Rückmeldungen (von Kunden)
- Konformität / Erfüllung der Produktanforderungen
- Prozess- und Produktmerkmale und deren Trends, einschließlich Möglichkeiten für Verbesserung
- Lieferanten
- Audits
- Seviceberichte, soweit angemessen
Im letzten Unterabschnitt der ISO 13485 geht es schließlich um die Verbesserung. Hier finden Sie Anforderungen an Korrektur- und Verbesserungsmaßnahmen. Einerseits benötigen Sie beschriebene Verfahren, mit denen Sie Abweichungen und Nichtkonformitäten in ihrem QM System und bei den Produkten wirksam beheben können. Des Weiteren müssen Sie Prozesse definieren, um auch potentielle Fehler – also solche, die noch nicht eingetreten sind – zu verhindern. Die Maßnahmen werden unter dem Begriff CAPA (Corrective and Preventive Action) zusammengefasst.
Welche Vorteile bietet eine ISO 13485 Zertifizierung?
Ein Qualitätsmanagementsystem nach ISO 13485 bietet zahlreiche Vorteile für Unternehmen, die Medizinprodukte entwickeln, herstellen oder vertreiben. Lesen Sie im folgenden, welche Vorteile sich durch die Einführung und Zertifizierung des QM-Systems ergeben können.
1. Erfüllung regulatorischer Anforderungen
Ein zentrales Ziel der ISO 13485 ist es, Unternehmen dabei zu unterstützen, die gesetzlichen und behördlichen Anforderungen für Medizinprodukte zu erfüllen. In vielen Ländern ist die Zertifizierung nach ISO 13485 eine Voraussetzung, um Produkte überhaupt auf den Markt zu bringen. Ein QM-System nach dieser Norm hilft Unternehmen, die Einhaltung von Vorschriften wie der Europäischen Medizinprodukteverordnung (MDR) oder der FDA-Regularien in den USA sicherzustellen. Dies reduziert das Risiko von Marktzulassungsproblemen und rechtlichen Konsequenzen.
2. Erhöhte Produktsicherheit und -qualität
Durch die Umsetzung der ISO 13485 Anforderungen wird sichergestellt, dass alle Prozesse im Unternehmen auf die Sicherheit und Qualität der Medizinprodukte ausgerichtet sind. Die ISO 13485 Inhalte zielen darauf, dass Unternehmen systematisch Risiken bewerten, qualitätsrelevante Prozesse kontrollieren und alle Anforderungen an das Produkt einhalten. Dies führt zu einer konsistenten Produktqualität und minimiert das Risiko von Produktfehlern, was wiederum das Vertrauen der Kunden in die Produkte stärkt.
3. Verbesserte Prozesskontrolle und Effizienz
Ein QM-System nach ISO 13485 hilft Unternehmen, ihre internen Prozesse besser zu strukturieren und zu steuern. Durch klar definierte Abläufe und Verantwortlichkeiten können Fehler frühzeitig erkannt und korrigiert werden, bevor sie zu größeren Problemen führen. Dies führt nicht nur zu einer verbesserten Qualität der Produkte, sondern auch zu einer höheren Effizienz im gesamten Produktionsprozess. Durch die kontinuierliche Überwachung und Optimierung der Prozesse können Unternehmen Ressourcen effizienter nutzen, Kosten senken und Produktionszeiten verkürzen.
4. Kundenzufriedenheit und Marktvertrauen
Die Zertifizierung nach ISO 13485 zeigt Kunden und Partnern, dass das Unternehmen höchsten Wert auf Qualität und Sicherheit legt. Dies kann einen Wettbewerbsvorteil bieten, da die Zertifizierung das Vertrauen in die Produkte erhöht und als Zeichen für Verlässlichkeit und Professionalität wahrgenommen wird. Kunden wissen, dass die Produkte strengen Qualitätsstandards entsprechen und dass das Unternehmen in der Lage ist, auf Probleme schnell und effektiv zu reagieren.
5. Risikominimierung mit der ISO 13485
Die Inhalte der ISO 13485 sind darauf ausgerichtet, ein systematisches Risikomanagement durchzuführen, um potenzielle Gefahren für Patienten und Anwender zu minimieren. Das bedeutet, dass Risiken nicht nur während der Produktentwicklung, sondern auch während der Herstellung, Lieferung und Anwendung regelmäßig überprüft und überwacht werden müssen. Durch diese proaktive Vorgehensweise können Unternehmen potenzielle Probleme frühzeitig erkennen und Maßnahmen ergreifen, um das Risiko von Produktfehlern oder Rückrufen zu minimieren.
6. Kontinuierliche Verbesserung
Ein weiterer wesentlicher Vorteil eines QM-Systems nach ISO 13485 ist der Fokus auf die kontinuierliche Verbesserung. Unternehmen sind verpflichtet, regelmäßig ihre Prozesse, Produkte und Dienstleistungen zu überprüfen und nach Wegen zu suchen, um diese zu optimieren. Dies fördert eine Kultur der ständigen Weiterentwicklung und Innovation, was sowohl der Qualität der Produkte als auch der Effizienz des Unternehmens zugutekommt.
7. Verbesserte Kommunikation und Zusammenarbeit
Die Norm fördert eine klare und transparente Kommunikation innerhalb des Unternehmens sowie mit externen Partnern und Lieferanten. Dokumentierte Verfahren und definierte Verantwortlichkeiten stellen sicher, dass alle Beteiligten auf dem gleichen Stand sind. So werden mögliche Missverständnisse oder Fehler reduziert. Die Anforderungen an das Lieferantenmanagement verbessern auch die Zusammenarbeit mit Lieferanten, was zu einer stabileren Lieferkette und einer höheren Qualität der eingekauften Materialien führt.
Wie ist der Ablauf einer ISO 13485 Zertifizierung?
Die ISO 13485 Zertifizierung ist ein international anerkanntes Zertifikat für Qualitätsmanagementsysteme in der Medizinbranche. Der Ablauf einer ISO 13485 Zertifizierung kann grob in folgende Schritte unterteilt werden:
- Vorbereitung: Die Vorbereitung auf eine ISO 13485 Zertifizierung umfasst die Identifikation der relevanten Anforderungen, die Überprüfung der aktuellen Systemdokumentation, die Durchführung von internen Audits und die Schulung der Mitarbeiter. Es ist auch wichtig, ein Zertifizierungsunternehmen auszuwählen, das die Zertifizierung durchführen wird.
- Vorprüfung: Eine Vorprüfung, auch als Gap-Analyse bezeichnet, kann durchgeführt werden, um Schwachstellen im bestehenden Qualitätsmanagementsystem zu identifizieren und zu beheben. Dadurch kann sichergestellt werden, dass das Unternehmen bereit ist, den Zertifizierungsprozess zu beginnen.
- Zertifizierungsaudit: Das Zertifizierungsaudit ist die Hauptprüfung, die von einem unabhängigen Zertifizierungsunternehmen durchgeführt wird. Das Audit besteht aus zwei Phasen. In der ersten Phase werden die Systemdokumentation und das Qualitätsmanagementsystem geprüft, um sicherzustellen, dass alle Anforderungen der ISO 13485 erfüllt sind. In der zweiten Phase werden die tatsächlichen Prozesse und Verfahren des Unternehmens geprüft, um sicherzustellen, dass sie den Anforderungen der ISO 13485 entsprechen.
- Bericht und Zertifikat: Nach Abschluss des Zertifizierungsaudits wird dem Unternehmen ein Auditbericht überreicht. Wenn alle Anforderungen erfüllt sind, wird das Unternehmen offiziell zertifiziert und erhält ein ISO 13485 Zertifikat.
- Überwachungsaudits: Um sicherzustellen, dass das Unternehmen weiterhin den Anforderungen der ISO 13485 entspricht, werden regelmäßige Überwachungsaudits durchgeführt. Diese Audits werden in der Regel einmal jährlich durchgeführt.
- Rezertifizierung: Alle drei Jahre muss das Unternehmen eine Rezertifizierung durchführen, um sicherzustellen, dass es weiterhin den Anforderungen der ISO 13485 entspricht.
Es ist wichtig zu beachten, dass der genaue Ablauf einer ISO 13485 Zertifizierung je nach Zertifizierungsunternehmen und Land, in dem das Unternehmen ansässig ist, variieren kann.
Welche weiteren Vorschriften und Normen gibt es für das Medizinprodukte Qualitätsmanagement?
Zahlreiche nationale sowie internationale Vorschriften und Normen stellen Anforderungen an das Qualitätsmanagement für Medizinprodukte. Für Europa sind hier die ISO 13485 , die EU-Medizinprodukteverordnung (MDR) oder auch die ISO 14971 von besonderer Relevanz. Die MDR fordert von Medizinprodukteherstellern ein zertifiziertes Qualitätsmanagement System. Bei der ISO 13485 handelt es sich um die dazu harmonisierte Norm. Sie beschreibt dabei Anforderungen an ein Qualitätsmanagementsystem. Um die Zertifizierung zu erhalten, muss das QM System diese Anforderungen zwingend erfüllen. Außerhalb Europas, zum Beispiel in den USA, gelten spezifische regulatorische Anforderungen. Die amerikanische „Food and Drug Administration“ (FDA) hat beispielsweise die „Quality System Regulation“ veröffentlicht. Für den chinesischen Markt ist die National Medical Products Administration (NMPA) für die Registrierung medizinischer Produkte verantwortlich.
Die EU-Medizinprodukteverordnung (MDR)
Mit der europäischen Medizinprodukteverordnung (MDR) wurden die Medizinprodukterichtlinie (MDD) und die Richtlinie über aktive implantierbare Medizinprodukte (AIMDD) ersetzt. Seit dem 25. Mai 2021 ist diese Ablösung offiziell. Allerdings stellt die praktische Umsetzung der Anforderungen aus der MDR für viele Hersteller eine große Herausforderung dar. Zum einen sind einige Voraussetzungen zu diesem Zeitpunkt noch nicht erfüllt, wie zum Beispiel eine Vielzahl noch nicht erlassener Rechtsakte auf die die MDR verweist. Zum anderen ist die Menge an Regeln, Gesetzen und Empfehlungen sehr groß. Daher stehen vor allem kleinere Unternehmen vor dem Problem die Anforderungen der MDR nicht erfüllen zu können.
Im Folgenden haben wir für Sie die wichtigsten Änderungen der Anforderungen der MDR im Vergleich zur (AI)MDD aufgelistet:
- Forderungen eines Sicherheitskonzepts, welches sich am kompletten Lebenszyklus eines Produkts orientiert
- die Anforderungen an benannte Stellen wurden erhöht
- der Anwendungsbereich ist größer und beinhaltet eine Vielzahl an Produkte die sich auf andere Medizinprodukte beziehen sowie Produkte ohne medizinischen Zweck
- ein neues System mit welchem Produkte identifiziert und zurückverfolgt werden können
- ein unabhängiges Expertengremium soll bei der Bewertung spezifischer Hochrisiko Produkte hinzugezogen werden
- die Hersteller sollen eine Vielzahl an Daten in die EUDAMED-Datenbank eingeben
- die Veräußerung von Medizinprodukten online unterliegt nun bestimmten Anforderungen
EU-Verordnung über In-vitro-Diagnostika (IVDR)
Die Abkürzung IVDR steht für „In Vitro Diagnostic Regulation,“ zu Deutsch „Verordnung über In-vitro-Diagnostika.“ Es handelt sich um eine Verordnung der Europäischen Union (EU), die die Herstellung, Vermarktung und Verwendung von In-vitro-Diagnostik-Geräten (IVDs) regelt. In-vitro-Diagnostik-Geräte sind solche, die zur Untersuchung von Proben aus dem menschlichen Körper, wie Blut, Urin oder Gewebe, bestimmt sind, um Informationen für medizinische Diagnosen, Prävention, Überwachung, Behandlung oder Linderung von Krankheiten bereitzustellen.
Die IVDR führt strengere Anforderungen an die Klassifizierung, klinische Evidenz, Leistungsbewertung und Post-Market-Überwachung von IVDs ein. Ziel ist es, die Sicherheit und Zuverlässigkeit dieser Geräte zu verbessern und gleichzeitig ihre ordnungsgemäße Verwendung und Effektivität im Gesundheitswesen sicherzustellen. Hersteller von IVDs müssen die Vorschriften der IVDR einhalten, um ihre Produkte weiterhin in der EU vermarkten zu können.
Welche Vorlagen, Musterdokumente und Checklisten benötigen Sie für Ihr Qualitätsmanagementsystem für Medizinprodukte?
Wir bieten Ihnen zahlreiche Checklisten und Mustervorlagen zum Qualitätsmanagement im Medizinproduktebereich an. Alle Vorlagen erhalten Sie in einem offenen Format, sodass Sie diese auf Ihre Bedürfnisse anpassen und direkt im Unternehmen anwenden können. Mehr Informationen finden Sie in unserer Rubrik der Musterdokumente zur ISO 13485.





Ich helfe Ihnen gerne weiter!
Kati Schäfer
Produktmanagement Training & PRO SYS
Tel.: 07231 92 23 91 - 0
E-Mail: kschaefer@vorest-ag.de
Unsere Serviceangebote im Qualitätsmanagement für Medizinprodukte
- Grundlagenwissen zum Thema: Qualitätsmanagement Medizinprodukte ISO 13485 – Definition & Ziele
- Ausbildungen & Weiterbildungen: ISO 13485 Schulung - Qualitätsmanagement Medizinprodukte
- E-Learning Kurse: Medizinprodukte Online Schulungen & ISO 13485 E-Learning
- Inhouse-Training: Qualitätsmanagement Medizinprodukte Inhouse Schulungen bei Ihnen im Unternehmen
- Musterdokumente: Qualitätsmanagement Medizinprodukte Vorlagen und Checklisten
- Wissensbausteine: Expertenwissen zum Qualitätsmanagement Medizinprodukte
- Fachzeitschrift PRO SYS: Monatliche Fachinfos inklusive Musterdokumente
- Beratung: Wir unterstützen Sie beratend zum Qualitätsmanagement Medizinprodukte
|
Seite 0 von 0
|