
Entwicklung von Medizinprodukten gemäß ISO 13485 und MDR 2017/745

zzgl. MwSt.
- Virtual Classroom-Preis: 616,55 € zzgl. MwSt.
- Dauer: 1 Tag (von 09:00 - 17:00 Uhr)
- Servicebausteine: im Wert von 74.80 €
- Artikelnummer: S232
- Max. Teilnehmer: 15
- Kursform: Präsenz oder Virtual Classroom
- Seminarinfo: PDF-Download
- Inhouse: Ihre unverbindliche Anfrage
-
Nächste Termine:
Live Virtual-Classroom-Training - 04.07. - 04.07.25. Bernd Stadter
Live Virtual-Classroom-Training - 04.11. - 04.11.25. Bernd Stadter
Alle Termine anzeigen
-
Weitere Kursformate:
Dieses Seminar zur Entwicklung von Medizinprodukten bereitet Sie optimal auf die Umsetzung der normativen und regulatorischen Anforderungen gemäß Abschnitt 7.3 der ISO 13485 sowie der EU Medizinprodukteverordnung vor. Der Prozess der Produktentwicklung spielt im Rahmen der Medizinprodukt Zulassung eine wichtige Rolle. Neben dem Medizinprodukterecht müssen Sie dabei noch weitere regulatorische und normative Anforderungen berücksichtigen. In unserer Schulung zur Entwicklung von Medizinprodukten lernen Sie, woher die Vorgaben stammen und wie Sie diese verstehen.
Außerdem lernen Sie in diesem Seminar die grundlegenden Sicherheits- und Leistungsanforderungen der Verordnung MDR 2017/745 kennen. Weiterhin erfahren Sie, welche Nachweise erstellt werden müssen und worauf Sie im Rahmen des Entwicklungsprozesses explizit achten müssen. Am Ende dieser Schulung können Sie die für den Prozess der Produktentwicklung geforderte Dokumentation erstellen und die allgemeinen Leistungs- und Sicherheitsanforderungen einhalten. Dabei bringt Ihnen Ihr Trainer das Wichtigste zum Thema näher, bereitet Sie auf mögliche Herausforderungen im Entwicklungsprozess vor und gibt Ihnen Anregungen zur praktischen Umsetzung.

Was sind die Inhalte in Ihrem Seminar zur Entwicklung von Medizinprodukten?
Lernen Sie in diesem Seminar alles Wichtige zur Umsetzung der regulatorischen sowie normativen Vorgaben der ISO 13485 und MDR im Rahmen des Entwicklungsprozesses von Medizinprodukten.
- Normativer Hintergrund für den Entwicklungsprozess in Europa mit der ISO 13485
- Regulatorische Hintergründe in Europa mit der MDR 2017/745/EU und IVDR 2017/746/EU
- Die Phase der Entwicklungsplanung sowie die Entwicklungseingaben
- Ergebnisse und Bewertung der Entwicklung des Produkts
- Verifizierung und Validierung
- Design Transfer in die Produktion
- Technische Dokumentation und Lenkung von Entwicklungsänderungen
- Entwicklungsakten
Welches Zertifikat und welche Qualifikationsbescheinigung erhalten Sie nach dieser Schulung?
Sie erhalten eine Qualifikationsbescheinigung zur Teilnahme an der Schulung in Deutsch sowie in Englisch. Die englische Qualifikationsbescheinigung ist dabei als Serviceleistung im Preis enthalten. Die Qualifikationsbescheinigung dokumentiert dabei die behandelten Inhalte sowie die Schulungsdauer und dient Ihnen als Nachweis Ihrer Teilnahme an der Schulung.


Was ist das Ziel in diesem Seminar zur Entwicklung von...
Welche Zielgruppe sprechen wir mit dieser Schulung...
Voraussetzungen für die Teilnahme an diesem Seminar zur...
Was ist das Ziel in diesem Seminar zur Entwicklung von Medizinprodukten nach ISO 13485?
Sie lernen in dieser Ausbildung die normativen sowie regulatorischen Anforderungen an alle Phasen im Prozess der Produktentwicklung kennen und erfolgreich umzusetzen. Ferner können Sie nach Abschluss der Schulung den Entwicklungsprozess gemäß den Vorgaben dokumentieren und alle Herausforderungen meistern.
Welche Zielgruppe sprechen wir mit dieser Schulung an?
Diese Schulung zur Entwicklung von Medizinprodukten richtet sich an Entwickler, Projektverantwortliche, Produktmanager, Beauftragte der Zulassung sowie alle anderen Personen, die für den Entwicklungsprozess verantwortlich sind.
Voraussetzungen für die Teilnahme an diesem Seminar zur Entwicklung von Medizinprodukten nach ISO 13485 und MDR
Für die Teilnahme an diesem Seminar sind Grundkenntnisse in der ISO 13485 (analog unserer Ausbildung Basiswissen ISO 13485) sowie zur MDR (analog unserer Schulung zur aktuellen EU Medizinprodukte Verordnung 2017/745) verausgesetzt. Einen Nachweis müssen Sie jedoch nicht erbringen. Die Anmeldung kann direkt vorgenommen werden.
Ihre Servicebausteine in diesem Entwicklung von Medizinprodukten gemäß ISO 13485 und MDR 2017/745 Kurs:
Als Kursteilnehmer erhalten Sie folgende Servicebausteine im Rahmen Ihrer Kursteilnahme. Diese Bausteine sind im Seminarpreis bereits enthalten und unterstützen Sie mit zusätzlichen Inhalten und Musterdokumenten zu Ihrem Kursthema. Den monatlichen Expertenbrief erhalten Sie erstmalig im Anschluss an Ihre Teilnahme. Dieser informiert Sie monatlich über aktuelle Fachinfos zu Ihrem Kursthema.
| Vorlagen: | Checkliste Übertragung der Entwicklung - Design Transfer Plan - Report Im Wert von 49,90 € |
|
| Vorlagen: | MDR 2020 - Leitfaden zur Umsetzung der EU Medizinprodukteverordnung MDR 2017/745/EU Im Wert von 24,90 € |
|
| Fachinfo: | Ihr monatlicher Expertenbrief Exklusive Expertentipps und Fachwissen für Sie |
Weitere mögliche Schritte Ihrer Ausbildung
Im Bereich Qualitätsmanagement in der Medizinprodukteindustrie haben wir zahlreiche Schulungen sowie modulare Ausbildungsmöglichkeiten für Sie im Angebot. Bilden Sie sich jetzt umfassend in Ihrem persönlichen Themengebiet weiter! Nachfolgend haben wir einige Schulungsmöglichkeiten für Sie zusammen gestellt.
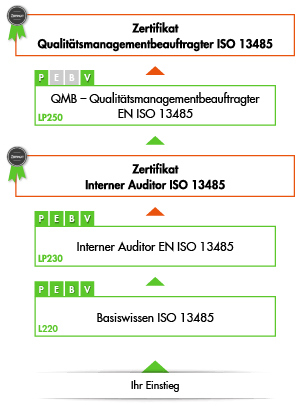
Ausbildung zum Qualitätsmanagementbeauftragten ISO 13485
Diese Ausbildung besteht aus drei Ausbildungsschritten bis hin zur Prüfung zum international anerkannten "Qualitätsmanagementbeauftragten ISO 13485". Sie können aber selbstverständlich auch jede Schulung einzeln besuchen, unabhängig von der Gesamtausbildung.
1. Schritt: Basiswissen Qualitätsmanagement ISO 13485
2. Schritt: Interner Auditor ISO 13485
3. Schritt: Qualitätsmanagementbeauftragter ISO 13485
Weiterbildungen gemäß der EU Medizinprodukte Verordnung - MDR 2017/745
Entdecken Sie weitere thematisch passende Schulungen rund um MDR und setzen Sie die Anforderungen erfolgreich in Ihrem Unternehmen um.
- Die aktuelle EU Medizinprodukte Verordnung - MDR 2017/745
- Medizinprodukte Vigilanz & Post Market Surveillance gemäß MDR 2017/745
- Technische Dokumentation für Medizinprodukte gemäß MDR 2017/745
- UDI Kennzeichnung für Medizinprodukte und grundlegende Sicherheits- und Leistungsanforderungen (GSPR)
- Medizinprodukte Händler und Importeure - Pflichten gemäß MDR Artikel 14 & 13
- ISO 14971 Risikomanagement für Medizinprodukte gemäß MDR 2017/745
- Online Schulung Verantwortliche Person MDR Artikel 15
- Schulung zum Medizinprodukteberater nach § 83 MPDG
|
Seite 0 von 0
|